
Lu Cheng
I am the Principal Investigator (Academy Research Fellow) leading the Computational Genomics research group at the Department of Computer Science, Aalto University. My main research interests are developing statistical and machine learnig methods for applications in single cell biology, metagenomics, epigenetics.
Email / Google Scholar / ORCID /
News
Research Projects
Biology has stepped into a new era where data are generated at an unprecedented pace. It is therefore necessary to develop new methods to analyse these ever increasing datasets. Thanks to the vast amount of data, researchers have a nice opportunity to come up with new biological hypotheses and look into the data from a new perspective. This kinds of projects may generate new statistical methods, new biological insights. In practice, we can make novel biological predictions and validate them in wet lab. I believe this type of data-driven research paradigm will become more and more popular in the future.
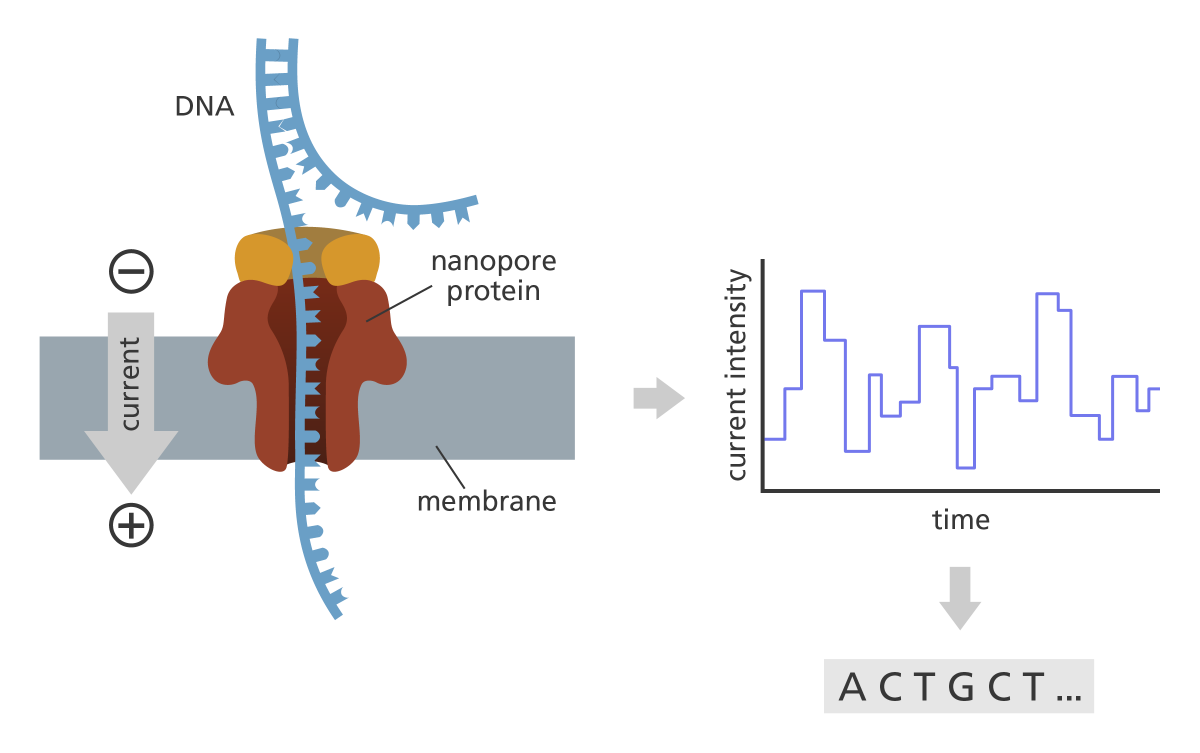
Oxford Nanopore (ONT) is the third generation sequencing technology. It measures the electric current when a RNA molecule passes through the pore. In principle, different signals are generated for different nucleotides. As we know, there exists different various modifications, e.g. m6A, in the naive RNA molecule. We aim to develop a Gaussain process based model to estimate these modifications from Nanopore data.
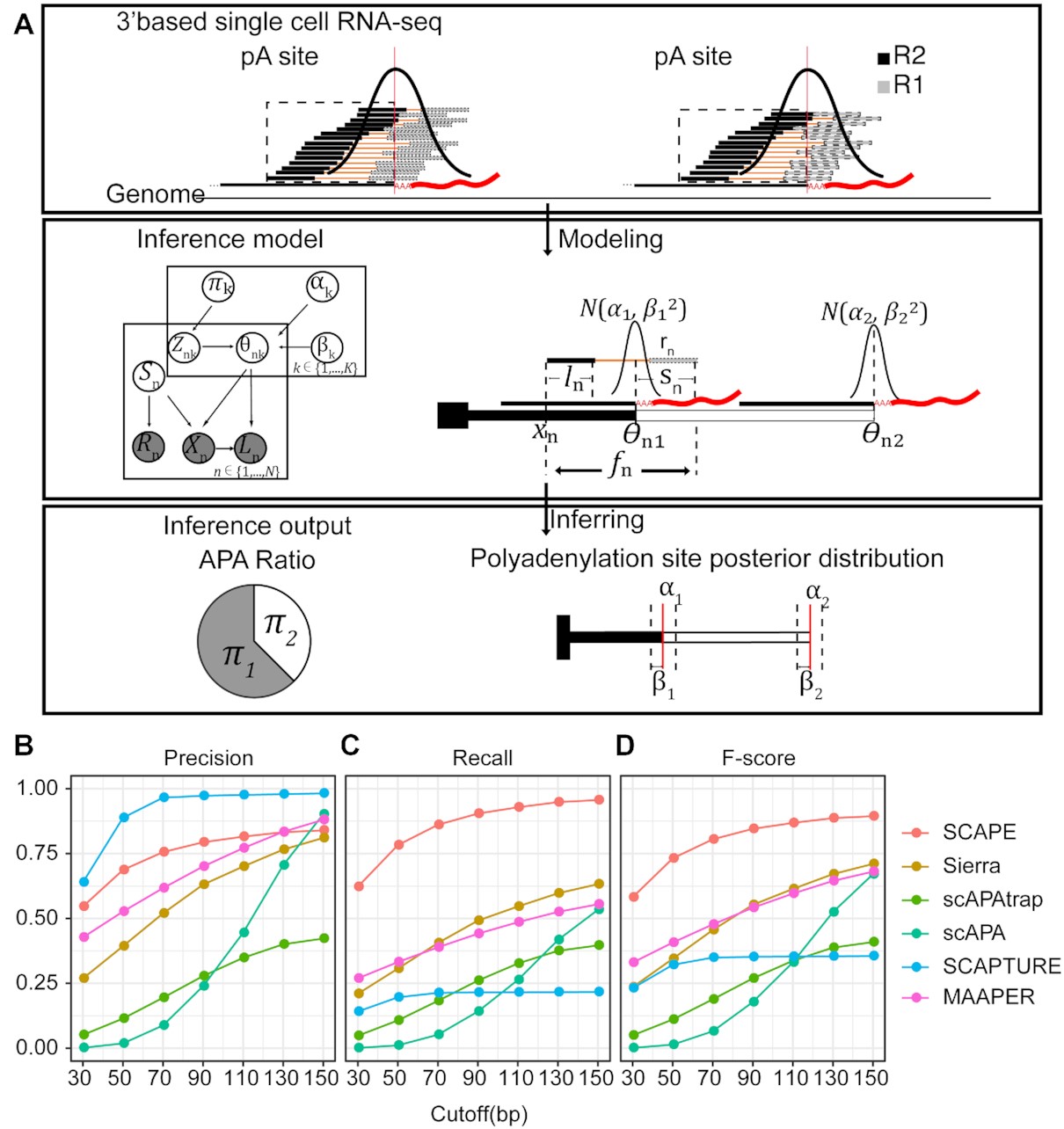
Alternative polyadenylation (APA) is one kind of alternative splicing in the 3'-UTR part of a gene. Common single cell RNA-seq (scRNA-seq) technologies such as 10x happen to measure the 3'-UTR of the gene, thus scRNA-seq data could be used to estimate APA events. We notice that the insert size information is standard in the sequencing library prepartion. This important prior information help us to develop an elegant Bayesian mixture model that shows superior performance than state of the art. Next, we demonstrated differential APA usage at different levels including organs, cell types, cell states (tumour or normal). The top differentiated APA events also show a strong association with miRNA expression. Furthermore, we demonstrated genome-wide dynamic changes of APA usage during stem cell differentiation (erythropoiesis and iPSC).
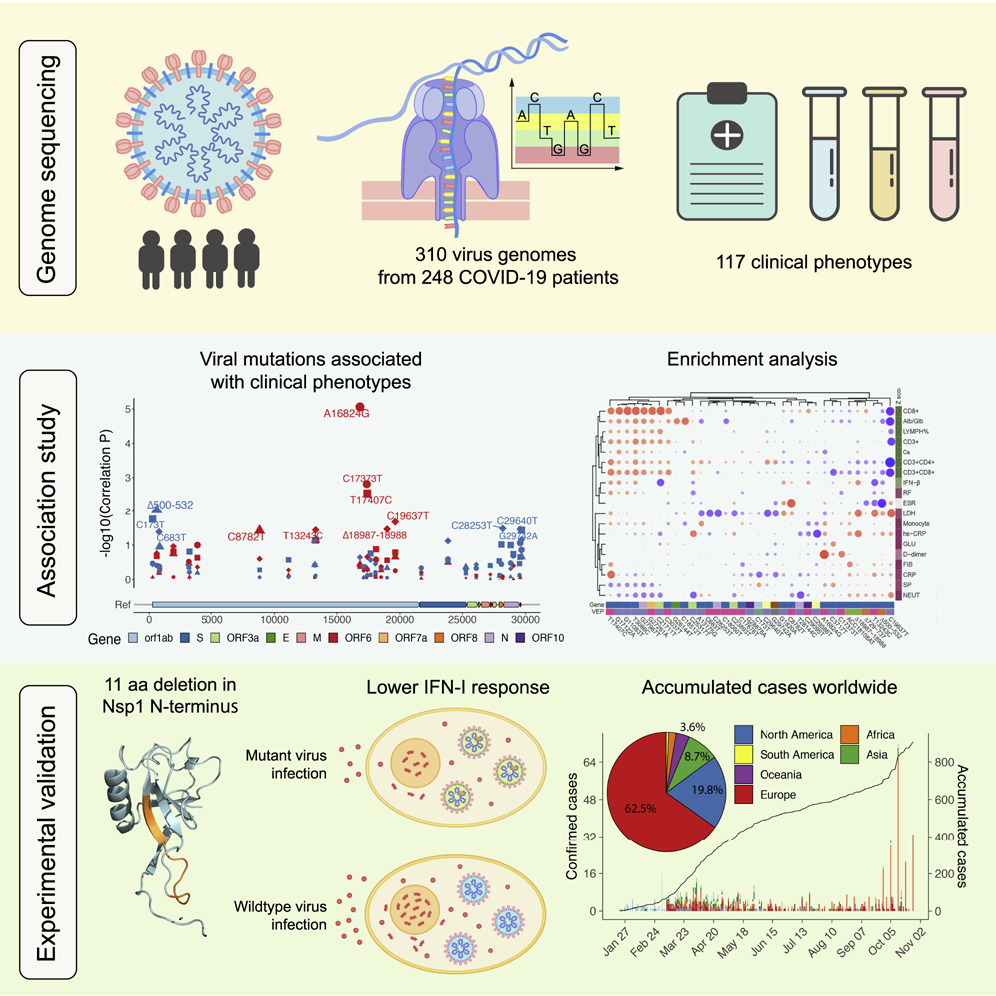
We have got hundreds of SARS-COV-2 genomes from COVID-19 diagnosed patients and their clinical data from our collaborators. We ask what are the key genotypes of the SARS-COV-2 genome that affect the symptoms of the patients. Through a GWAS study, we find a deletion in the nsp1 gene that is associated with the severeness of the symptoms. This deletion location turns out to be a hopspot in many different SARS-COV-2 strains worldwide. Then we show that the deletion induces with lower type I interferon response in wet lab.
 Project Page | Paper
Project Page | Paper
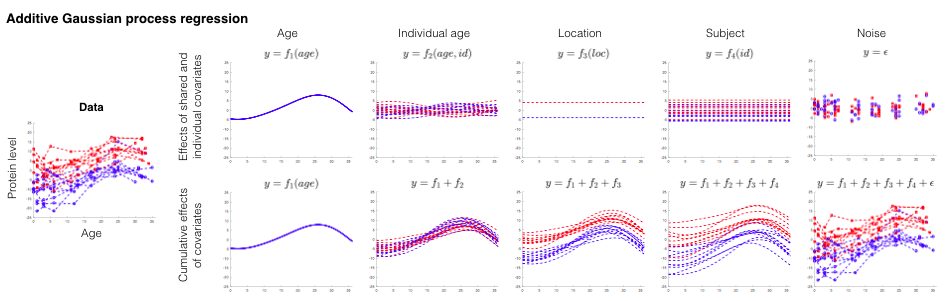
The study design of a biological experiment can be very complex, especially when collecting time series data from different individuals. Many different covariates in the longitudinal study design need to be considered. These covariates may affect the target variable in a nonlinear way. What are the important covariates and how do they affect the target variable? We developed a tool called LonGP to decompose the raw data into several linear/nonlinear functions of the covariates. Application of this method to a published longitudinal type 1 diabetes (T1D) proteomics dataset helps us to identify some proteins that show weak changes near the seroconversion event.
Selected publications
✲: (co-)first author; #: (co-)coresponding author. For a full list, please check my Google Scholar.
- SCAPE: a mixture model revealing single-cell polyadenylation diversity and cellular dynamics during cell differentiation and reprogramming
Ran Zhou*, Xia Xiao, Ping He, Yuancun Zhao, Mengying Xu, Xiuran Zheng, Ruirui Yang, Shasha Chen, Lifang Zhou, Dan Zhang, Qingxin Yang, Junwei Song, Chao Tang, Yiming Zhang, Jing-wen Lin#, Lu Cheng#, Lu Chen#
Nucleic Acids Research 2022 | paper - Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response
Jing-wen Lin*# , Chao Tang* , Han-cheng Wei* , Baowen Du* , Chuan Chen* , Minjin Wang* , Yongzhao Zhou* , Ming-xia Yu* , Lu Cheng* , Suvi Kuivanen* , Natacha S. Ogando , Lev Levanov , Yuancun Zhao , Chang-ling Li , Ran Zhou , Zhidan Li , Yiming Zhang , Ke Sun , Chengdi Wang , Li Chen , Xia Xiao , Xiuran Zheng , Sha-sha Chen , Zhen Zhou , Ruirui Yang , Dan Zhang , Mengying Xu , Junwei Song , Danrui Wang , Yupeng Li , ShiKun Lei , Wanqin Zeng , Qingxin Yang , Ping He , Yaoyao Zhang , Lifang Zhou , Ling Cao , Feng Luo , Huayi Liu , Liping Wang , Fei Ye , Ming Zhang , Mengjiao Li , Wei Fan , Xinqiong Li , Kaiju Li , Bowen Ke , Jiannan Xu , Huiping Yang , Shusen He , Ming Pan , Yichen Yan , Yi Zha , Lingyu Jiang , Changxiu Yu , Yingfen Liu , Zhiyong Xu , Qingfeng Li , Yongmei Jiang , Jiufeng Sun , Wei Hong , Hongping Wei , Guangwen Lu , Olli Vapalahti , Yunzi Luo , Yuquan Wei , Thomas Connor , Wenjie Tan , Eric J. Snijder , Teemu Smura# , Weimin Li #, Jia Geng #, Binwu Ying #, Lu Chen#
Cell Host & Microbe 2021 | paper - An additive Gaussian process regression model for interpretable non-parametric analysis of longitudinal data
Lu Cheng*#, Siddharth Ramchandran, Tommi Vatanen, Niina Lietzén, Riitta Lahesmaa, Aki Vehtari & Harri Lähdesmäki#
Nature Communications 2019 | paper - Hierarchical and spatially explicit clustering of DNA sequences with BAPS software
Lu Cheng*, Thomas R. Connor, Jukka Sirén, David M. Aanensen, Jukka Corander#
Molecular Biology and Evolution 2013 | paper - Bayesian estimation of bacterial community composition from 454 sequencing data
Lu Cheng*#, Alan W. Walker, Jukka Corander
Nucleic Acids Research 2012 | paper